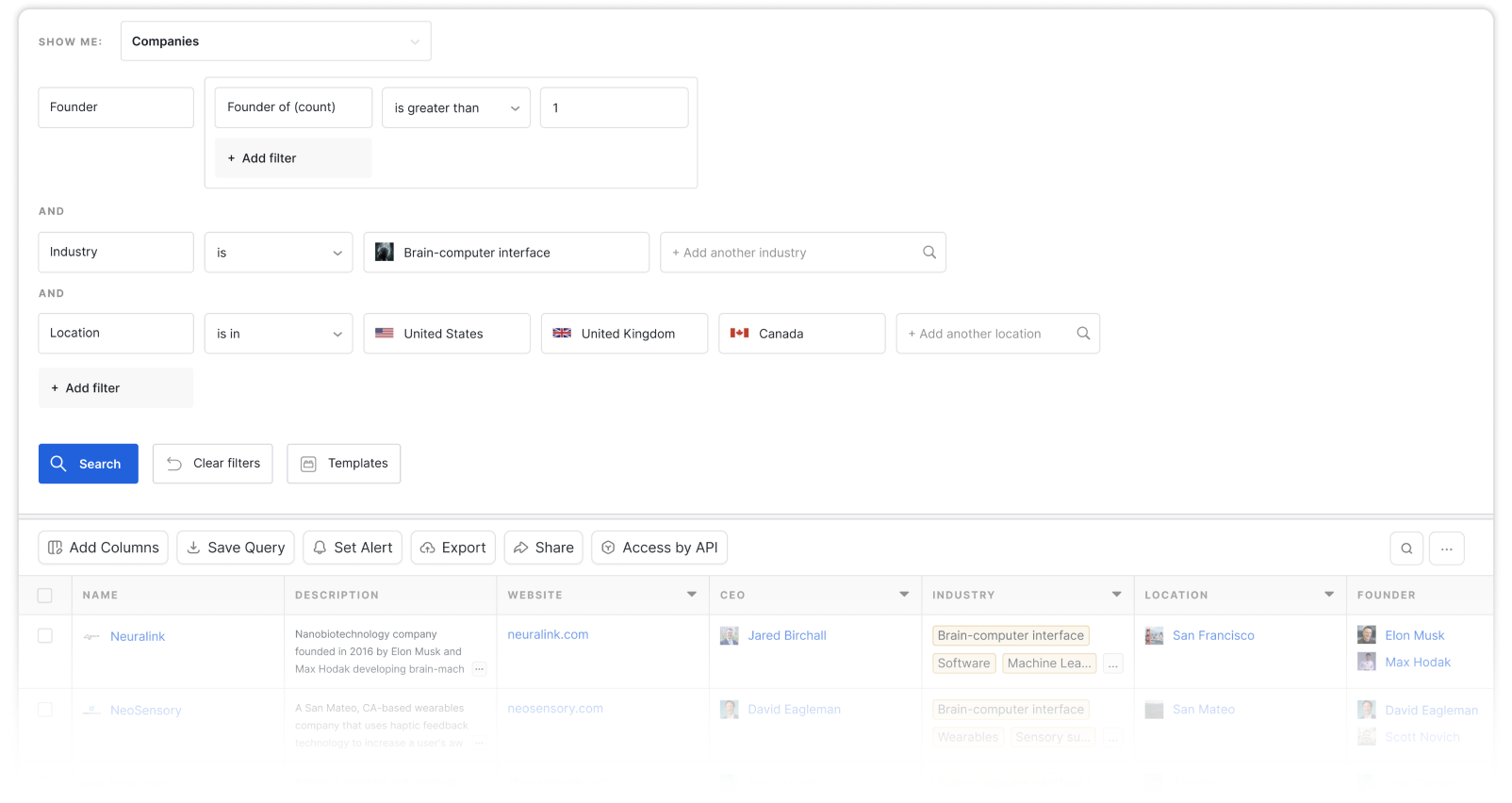
Bacterial Genome Sequencing
The determination of the sequence of nucleic acids in genomic DNA of bacteria using DNA sequencing techniques
All edits

Bacterial Genome Sequencing
The determination of the sequence of nucleic acids in genomic DNA of bacteria using DNA sequencing techniques
Bacterial genome sequencing is the determination of the sequence of nucleic acids in genomic DNA of bacteria using DNA sequencing techniques. The first bacterial genomes to have complete genomes sequencedsequences were Haemophilis influenze and Mycoplasm genitalium, published in 1995. By 2017 about 12,300 bacterial genomes had been sequenced. Bacterial genome sequences are publicly available at the NCBI Microbial Genomes page, the Genomes OnLine Database (GOLD) and Sequence Read Archive (SRA).
Bacterial genomes were mostly sequenced by the Sanger method prior to 2005 using whole genome shotgun libraries and capillary electrophoresis. This required assembly of sequences into a draft genome that was then finished. The cost of a finished bacterial genome was about $50,000. Next-Generation Sequencing (NGS), also known as second generation sequencing, allowed low-cost, high-throughput sequencing to be available to commercial vendors. In 2015 it was estimated that the cost of generating a draft bacterial genome can be less than $1. NGS technology such as Illumina produced shorter reads than Sanger sequencing which meant that the cost of the finishing steps, after the draft is complete, could be 95% of the total. Due to the cost of finishing, many genomes are published in draft status, in multiple contigs and varying quality.
Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. Pacbio RS II system can achieve complete genome assembly even in the regions of high or low GC content, as well as repetitive sequences. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data.
A closed, high-quality genome sequence for C. autoethanogenum DSM10061 was generated using only using PacBio sequencing to achieve "0 Gap" assembly and without the need for manual finishing. But there are still many gaps in the genome obtained using Illumina and 454 sequencing platforms. C.autoethanogenum and C. ljungdahliiare were indistinguishable at the 16S rRNA gene level and had high scores for similarity. Through whole-genome sequencing, it was found that there were significant differences in CRISPR system, hydrogenase and other aspects between the two, which were difficult to detect through second-generation sequencing.
The first bacterial genomes to have complete genomes sequenced were Haemophilis influenze and Mycoplasm genitalium, published in 1995. By 2017 about 12,300 bacterial genomes had been sequenced.

Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. Pacbio RS II system can achieve complete genome assembly even in the regions of high or low GC content, as well as repetitive sequences. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data. CD Genomics has completed hundreds of bacterial genome assembly cases without gap.

A closed, high-quality genome sequence for C. autoethanogenum DSM10061 was generated using only using PacBio sequencing to achieve "0 Gap" assembly and without the need for manual finishing. But there are still many gaps in the genome obtained using Illumina and 454 sequencing platforms. C.autoethanogenum and C. ljungdahliiare were indistinguishable at the 16S rRNA gene level and had high scores for similarity. Through whole-genome sequencing, it was found that there were significant differences in CRISPRCRISPR system, hydrogenase and other aspects between the two, which were difficult to detect through second-generation sequencing.

Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. Pacbio RS II system can achieve complete genome assembly even in the regions of high or low GC content, as well as repetitive sequences. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data. CD GenomicsCD Genomics has completed hundreds of bacterial genome assembly cases without gap.

Bacterial Genome Sequencing
CD Genomics provides bacterial whole genome sequencing with the PacBio Sequel system to help you rapidly advance your research to explore the bacterial genetic structure and functions.
CD Genomics is providing PacBio Single Molecular Real-Time (SMRT) sequencing to increase your research method for bacterial genome sequencing. A comprehensive view of the bacterial genome, including genes, regulatory regions, IS elements, phage integration sites, and base modifications is vital to understanding key traits such as antibiotic resistance, virulence, and metabolism.
At CD Genomics, we are using the long-read PacBio Sequel platform to support researchers all over the word with bacterial de novo whole genome sequencing needs. Our bioinformatics analysis include: Genome assembly and polishing, gene prediction, genome annotation, and comparative species genomes analysis.

Bacterial Genome Sequencing
CD Genomics provides bacterial whole genome sequencing with the PacBio Sequel system to help you rapidly advance your research to explore the bacterial genetic structure and functions.
CD Genomics is providing PacBio Single Molecular Real-Time (SMRT) sequencing to increase your research method for bacterial genome sequencing. A comprehensive view of the bacterial genome, including genes, regulatory regions, IS elements, phage integration sites, and base modifications is vital to understanding key traits such as antibiotic resistance, virulence, and metabolism.
At CD Genomics, we are using the long-read PacBio Sequel platform to support researchers all over the word with bacterial de novo whole genome sequencing needs. Our bioinformatics analysis include: Genome assembly and polishing, gene prediction, genome annotation, and comparative species genomes analysis.
Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. Pacbio RS II system can achieve complete genome assembly even in the regions of high or low GC content, as well as repetitive sequences. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data. CD Genomics has completed hundreds of bacterial genome assembly cases without gap.
A closed, high-quality genome sequence for C. autoethanogenum DSM10061 was generated using only using PacBio sequencing to achieve "0 Gap" assembly and without the need for manual finishing. But there are still many gaps in the genome obtained using Illumina and 454 sequencing platforms. C.autoethanogenum and C. ljungdahliiare were indistinguishable at the 16S rRNA gene level and had high scores for similarity. Through whole-genome sequencing, it was found that there were significant differences in CRISPR system, hydrogenase and other aspects between the two, which were difficult to detect through second-generation sequencing.
Bacterial Genome Sequencing
The determination of the sequence of nucleic acids in genomic DNA of bacteria using DNA sequencing techniques

Find more entities like Bacterial Genome Sequencing